
Highly sensitive & specific padlock probe chemistry
Only commercial in situ platform to use a padlock probe rolling circle amplification chemistry. This powerful approach allows:
- High sensitivity across a broad range of expression, including low expressing transcripts.
- Recognition of short or highly degraded transcripts (i.e. FFPE) without requiring a large number of probes per gene.
- Tunable sensitivity so that highly expressed transcripts can be included with minimal impact on performance.
- The ability to distinguish between transcripts with high homology, such as isoforms.
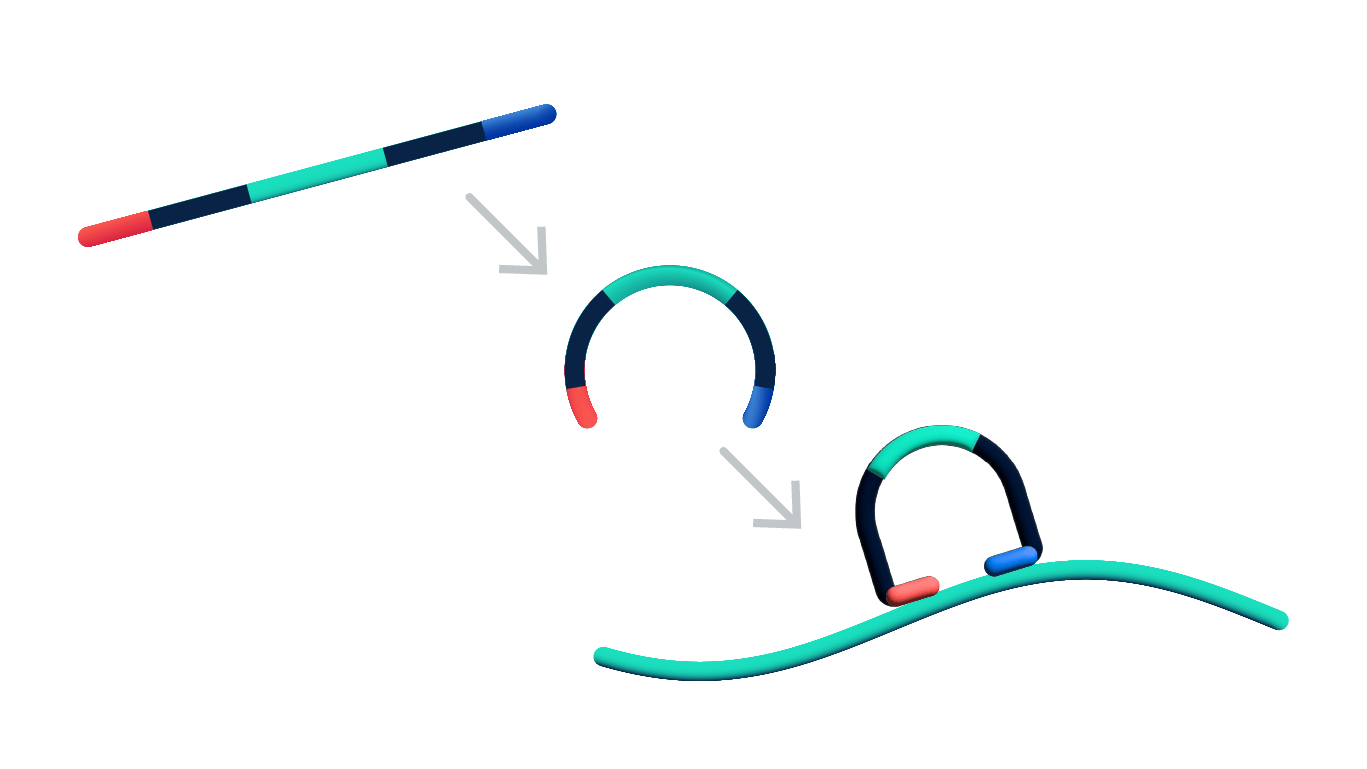
How it works
Once tissue sections are prepared on Xenium slides the chemistry workflow consists of probe hybridization, ligation, and enzymatic amplification. It is a unique four-step authentication process that is the key to Xenium’s high performance.
Steps 1 & 2: Dual target recognition & hybridization
Both probe arms must stably hybridize to their target. If only one arm hybridizes, the probe is unstable and will be washed off in the post hybridization wash. The padlock probes also contain a gene-specific barcode sequence that is used to generate a unique optical signature of each transcript.
Step 3: Probe ligation
Only when both probe arms are stably hybridized to the target with a perfect match does proximal probe ligation occur. Partially bound or mis-matching probes are not ligated and cannot be amplified.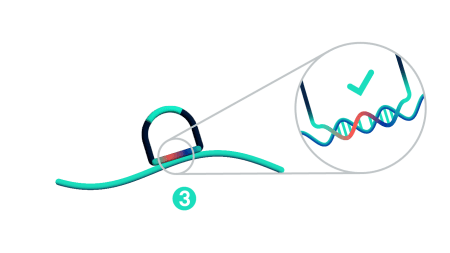
Step 4: Rolling circle amplification (RCA)
Only ligated probes are amplified, enriching for true target recognition events and suppressing off-target events. The RCA approach creates a robust and strong signal allowing single probe detection.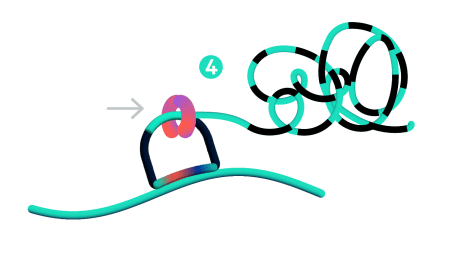
On-instrument detection & decoding
After the off-instrument chemistry workflow is completed the slides are loaded onto the Xenium Analyzer for transcript detection. The amplified products in the samples undergo successive rounds of fluorescent probe hybridization, imaging, and removal. An optical signature specific to each gene is generated, enabling identification of the target gene. Finally, a spatial map of the transcripts is built across the entire tissue section.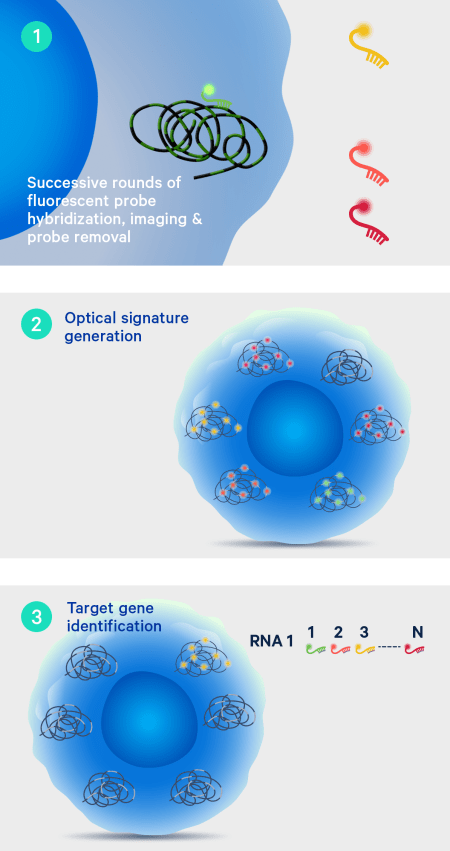
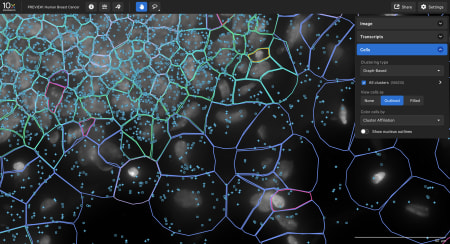
Explore the data
- The Xenium In Situ platform processes data in parallel to a run so the output can be directly explored after the instrument run. Sign up to get a personalized data demo exploring a human breast cancer or mouse brain Xenium In Situ dataset for yourself.
The end-to-end Xenium platform

Xenium In Situ Analyzer
Simple design and interface plus fast run times and on-instrument analysis combine for world-class in situ profiling.

Xenium In Situ reagents and panels
Choose from a diverse menu of curated panels, customizable to fit your research needs.

Data visualization
Xenium Explorer provides off-instrument interactive visualization, simplified data QC, and exploration of subcellular readouts.

Dedicated team of spatial experts
Get the help you need from our expert support team, reachable by phone or email.